藻资讯丨我国科研团队组装的造礁石珊瑚基因组被NCBI评选为主要标准基因组

至2024年10月,NCBI (National Center for Biotechnology Information)已公布了上百个造礁石珊瑚(Scleractinian corals)基因组[1],其中组装到染色体水平的基因组共有36个【桑格研究所(Wellcome Sanger Institute)提供了28个】。在这36个基因组中,经过NCBI多轮评估,共有29个被评选为参考基因组(Reference genome),但仅有5个被进一步评选为注释基因组【NCBI RefSeq (Reference Sequence Database);即标准基因组,俗称旗舰基因组】[2],作为全球石珊瑚研究的基因组背景。在这5个注释基因组中,有4个是来自于东南大学数字医学工程全国重点实验室陆祖宏、何春鹏课题组的组装成果。
从2022年开始,全球动植物基因组研究进入到了颠覆性变革时代。Wellcome Sanger Institute依靠PacBio HiFi (High-Fidelity)和Hi-C (High-throughput Chromosome Conformation Capture)联用技术系统【部分结合ONT (Oxford Nanopore Technologies)测序技术】率先发起“总攻”,对上万种重要的动植物基因组进行了地毯式的染色体水平测序组装(很多组装到了单倍型水平),涵盖了系统发生树(Phylogenetic tree)的所有重要节点,几乎占领了所有的NCBI参考基因组[1]和注释基因组[2]高地,迅速替代了大部分早期基于单一illumina二代测序技术所获得的基因组数据,在种质资源方面获得了空前的基因数据“大盘”优势。
但与普通陆生动物不同,石珊瑚等刺胞动物具有与虫黄藻等共生生物终生共生的特点,而且一种石珊瑚还经常与多种虫黄藻同时内共生[3],因此石珊瑚基因组在DNA提取方面属于典型的高污染基因组。再者,石珊瑚等海洋生物的基因组具有父母亲本高杂合的特性。以上两特性导致石珊瑚基因组是动物界中最难准确组装到染色体水平的基因组之一。新兴的HiFi测序技术虽然高效,但并不适合高污染、高杂合基因组的直接组装。虽然HiFi测序数据的准确性在PacBio CLR (Continuous Long Reads)测序数据的基础上提高了很多,但对于高污染或高杂合样本,HiFi测序数据在直接组装过程中会显著增加将污染或杂合序列组装进基因组的几率。从2017年开始,陆祖宏、何春鹏及课题组的10多名科研人员针对石珊瑚基因组的特点和PacBio、Nanopore及Illumina等高通量测序技术的各自优势,制定出一套基于初评、测序、排污、组装、注释的多步技术方案,分离、排除了多种共生藻的干扰,最终得到了组装质量较高的染色体水平的石珊瑚基因组。
陆祖宏、何春鹏课题组此次组装的4种石珊瑚基因组分别为:美丽鹿角(轴孔)珊瑚(Acropora muricata, GCF_036669905.1)、叶形表孔珊瑚(Montipora foliosa, , GCF_036669935.1)、叶板蔷薇珊瑚(Montipora capricomis, GCF_036669925.1)和疣状杯形珊瑚(Pocillopora verrucosa, GCF_036669915.1)。以上4种石珊瑚均为印度洋-太平洋地区(Indo-Pacific region)珊瑚礁中的主要优势物种,其中A. muricata、M. foliosa和P. verrucosa来源于西沙岛礁,为当地的常见物种。Acropora是石珊瑚目(Scleractinia)的第一大属,A. muricata是该属的常见优势种。Montipora是石珊瑚目的第二大属,M. foliosa是该属的常见优势种,M. capricomis则因其绚丽的颜色而在石珊瑚水族箱养殖产业中非常受欢迎。现生石珊瑚目可以分为Complexa和Robusta两大分支,Acropora和Montipora属于Complexa分支,Pocillopora属于Robusta分支。Acropora和Montipora两属对于维持珊瑚礁表面活体层的碳酸钙结构及生产能力至关重要。P. verrucosa是重要的礁前珊瑚,其分支粗壮,生长快速,个体直径大,具有形成生物水下消波块的能力。因此,这三个属的石珊瑚对生物造礁、护礁、固礁至关重要,是海洋建设土交材建工程的良好补充,是实现海洋碳中和与负碳排放的中坚力量。
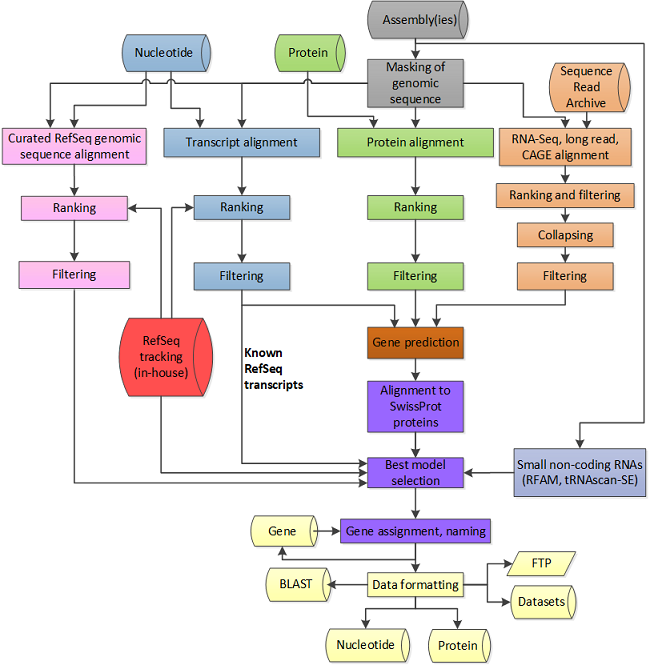
图1. NCBI标准化基因组注释流程[2]
RefSeq是由NCBI创建的一个高质量注释基因组数据库[图1]。NCBI对基因组是否能够被纳入RefSeq数据库有明确的筛选和评估标准。以下是能够获得NCBI注释,成为RefSeq基因组的一些关键条件:
1. 高质量的基因组组装结果
完整性:基因组必须具备较高的完整性,尽可能覆盖整个基因组,避免大量的缺失或未解析区域。
连续性:基因组序列应当具有较高的连续性,较少的片段化(Scaffolding),并且优选染色体级别的组装。
低错误率:基因组序列应尽量少有拼接错误或测序错误,通常需要经过多次校对和纠正。
覆盖深度:测序覆盖深度应该足够高,以确保基因组序列的准确性和完整性。
2. 物种的科学价值
医学、农业或环境重要性:与人类健康、农业、生态环境相关的重要物种通常优先获得注释。
模式生物:常用作研究对象的模式生物(如小鼠、斑马鱼、拟南芥等)往往被优先考虑,因为这些基因组对科学研究具有广泛的影响。
基因组学研究的基础性物种:对于特定分类群或进化研究具有代表性的物种,尤其是某些物种的基因组首次测序,通常具有较高的优先级。
3. 基因功能注释潜力
已知基因注释:基因组中包含已知基因或功能域,这些信息可以通过自动化或人工的方式进行准确注释。
功能基因预测:基因组需要具有较好的基因预测和功能域注释潜力,以便研究人员能够进行基因功能研究。
4. 测序技术和组装方法
先进的测序技术:基因组序列通常采用高通量测序技术生成,如PacBio或Nanopore测序,能够产生高质量的长读长数据。
优化的组装方法:基因组需要使用先进的组装算法,确保序列的准确拼接,并生成高质量的组装结果。
5. 基因组的公共可用性
公开数据提交:基因组数据必须提交到NCBI等公共数据库,并确保科学界可以免费访问和使用。研究者应将基因组数据提交至NCBI的GenBank或其他相关数据库,确保数据的公开性。
元数据完整:基因组应附有完整的元数据信息,包括物种分类信息、测序平台、测序深度、基因组大小、组装版本等。
6. 与参考标准的比对
物种代表性:如果某个物种的基因组被选择为RefSeq基因组,通常需要与其他物种的参考基因组进行比对和分析,以确保其准确性和生物学意义。
多样性基因组的纳入:对于某些物种,多个基因组版本可能会被注释为RefSeq基因组,代表该物种的不同个体或亚种,反映遗传多样性。
7. 符合RefSeq的政策和标准
RefSeq有一套严格的政策和标准,特别是在注释生物基因组时。这些标准涵盖基因组的质量、物种的科学意义、测序和组装的技术规范等。
NCBI RefSeq为研究人员提供了标准化、经过注释和功能标注的参考基因组序列,具有广泛的生物物种覆盖,不仅涵盖了人类基因组,还覆盖了大量的模式生物、病原体、农业重要物种等,支持广泛的基础和应用研究。其主要作用和数据优势包括:
主要作用
1. 基因组参考标准:RefSeq提供了高质量、经过人工和自动校对的基因组序列,作为生物基因组研究的标准参考。这为研究人员提供了一个可靠的基准,用于基因组比较、基因注释和功能研究。
2. 功能注释:RefSeq为基因组序列中的基因、转录本和蛋白质提供详细的功能注释,帮助科学家理解基因功能、调控机制以及与疾病的关系。
3. 跨物种比较:RefSeq不仅涵盖人类基因组,还包括植物、动物、微生物等多种物种的基因组数据,支持跨物种基因组比较分析。
4. 疾病研究:RefSeq在医学研究中至关重要。它帮助科学家识别与人类疾病相关的基因变异,为药物开发、基因疗法和个体化医疗提供了基础数据。
5. 数据整合与共享:RefSeq与其他NCBI数据库(如GenBank、dbSNP等)整合,便于研究人员获取不同类型的生物数据,从而实现跨平台的数据分析。
数据优势
1. 高质量和标准化:RefSeq中的基因组数据经过严格的质量控制和手动校正,确保数据的准确性和可靠性。相比其他数据库,RefSeq具有更高的标准化水平。
2. 持续更新:RefSeq定期更新,确保最新的基因组数据和注释反映最新的科学发现和研究成果。
3. 数据的可重复性和可追溯性:通过RefSeq,研究人员可以获取标准化的参考序列,从而保证研究结果的可重复性。每个参考序列都有唯一的标识符,便于追溯和引用。
4. 易于访问和使用:NCBI提供的各种工具(如BLAST、Genome Browser等)可以轻松地访问和分析RefSeq数据,方便研究人员进行查询和比对。
5. 免费公开获取:RefSeq数据免费对公众开放,任何研究人员都可以访问和下载这些数据,促进了全球范围内的科学研究和合作。
6. 推进生物技术和基因工程:高质量的基因组注释可以促进基因工程和生物技术的进步,例如通过基因编辑或基因合成等技术,进行生物制造、药物研发或环境修复等应用。
总之,当一个物种的基因组获得NCBI RefSeq注释后,将具有更加广泛的科学、医学和应用价值。它提供了高质量的基因组参考,支持深入的基因功能分析和跨物种比较研究。同时,通过标准化的数据格式和持续更新,帮助提高研究效率,促进国际合作,并推动在医学、农业和生物技术领域的应用。这些作用、优势和好处使得NCBI RefSeq成为基因组学领域的重要资源。
对于目前全球共生刺胞动物尤其是石珊瑚基因组的研究进展,何春鹏认为:虽然在国际科学界的共同努力下,取得了一定进展,获得了一定数量的组装质量相对较高的染色体水平基因组,但与小鼠、果蝇等模式生物和水稻、小麦等重要经济作物相比,还存在一定差距,如亲本单倍型基因组组装和基因水平转移(Horizontal Gene Transfer, HGT)现象确认等。古生物研究表明,石珊瑚与虫黄藻之间的内共生关系至少从开始进行生物矿化之初,即在三叠纪安妮期就已经形成[4]。科学界在几十年前就确信,经过至少2.4亿年的漫长演化史,石珊瑚和虫黄藻之间存在一定的基因水平转移现象,即有部分虫黄藻基因被水平转移到了石珊瑚基因组中,造成石珊瑚具有一定的植物特性[5]。但相关现象一直没有被准确的测序结果所证实,相关数据在上传NCBI过程中一直得不到官方认可,被作为污染序列直接屏蔽或删除。NCBI相关业务线负责人Linda Frisse博士认为,必须有更加准确的长读长测序数据(Reads),如基于read N50≥200 Kb的高覆盖度长读长测序数据组装出来的T2T (Telomere-to-Telomere)水平基因组(Contig N 50≥10 Mb),甚至是单倍型基因组,才能更好的准确解释相关现象。基于上述原因,Nature杂志资深编辑Michelle Trenkmann认为,尽管与十几年前相比,相关领域的研究已经取得了巨大进展,但还未能彻底解决石珊瑚基因组的底层问题,仍然需要进一步的“硬”创新。目前,HiFi测序技术全面替代CLR测序技术是大势所趋,超长DNA提取技术和超长ONT测序技术也在高速发展【基于早期ONT测序技术组装出来的石珊瑚基因组[6]和其他共生刺胞动物基因组(如八放软珊瑚基因组)并不成功[7]】。因此,在现有成果基础上,首先对HiFi reads进行排污,然后与超长ONT reads进行混合组装,再结合Hi-C技术进行辅助组装,有望快速获得T2T水平的高精度石珊瑚基因组[8]。在此基础上,既可以进一步探讨石珊瑚与共生藻之间的基因水平转移、石珊瑚单倍型基因组等之前难以有效探索的生物学问题,又可以基于AI (Artificial Intelligence)生物制药思维,依托新兴的H100等GPU平台和单细胞测序数据,对石珊瑚进行更加深入的蛋白结构重建(AlphaFold结合冷冻电镜技术)和细胞通讯(Cell chat)分析,找到与石珊瑚抗热、生长及其他抗胁迫相关的关键生物学因素,运用细胞目穿透转运和海洋缓释技术予以干涉,增加珊瑚礁生态修复的技术效果。
对于动植物基因组的基因注释信息获取和更新,陆祖宏认为:目前仍过度依赖NCBI平台,我国具有庞大的物种资源和生物信息分析需求,我国应加速发展自身的高通量长片段测序和基因注释平台,建立自己的测序数据存储和评价数据库。
该项目在执行过程,在石珊瑚的基础生物学特性、核型分析、生态分布等方面得到了广州海洋实验室、中国科学院南海海洋研究所张偲院士团队、张跃环研究员团队的大力指导和支持。
项目主要参与人员:陆祖宏、何春鹏、韩婷玉、黄万龙、王冰、张偲、张跃环、陈均远、廖馨、李一鑫、郭卓君、刘云卿、毕长伟、鲁娜。

参考文献
1. https://www.ncbi.nlm.nih.gov/datasets/genome/?taxon=6125
2. https://www.ncbi.nlm.nih.gov/refseq/annotation_euk/process/
3. Chen, B., Yu, K., Liang, J., Huang, W., Wang, G., Su, H., Qin, Z., Huang, X., Pan, Z., Luo, W., Luo, Y., & Wang, Y. (2019). Latitudinal Variation in the Molecular Diversity and Community Composition of Symbiodiniaceae in Coral From the South China Sea. Frontiers in microbiology, 10, 1278.
4. Stanley G. D., Jr (1981). Early history of scleractinian corals and its geological consequences. Geology, 9(11), 507–511.
5. Stanley G. D., Jr (2006). Ecology. Photosymbiosis and the evolution of modern coral reefs. Science, 312(5775), 857–858.
6. https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_041430625.1/
7. Hu, M., Zheng, X., Fan, C. M., & Zheng, Y. (2020). Lineage dynamics of the endosymbiotic cell type in the soft coral Xenia. Nature, 582(7813), 534–538.
8. Hu, Y., Zhang, Z., Sun, S., Sun, Y., Huang, H., Zhou, W., & Wei, F. (2024). Toward the generation of pure coral genomes with experimental and bioinformatic improvements. Innovation, 5(4), 100643.
www.algaehub.cn
admin@algaehub.cn
长按扫码访问Algae-Hub网站


如需了解Algae-Hub研修班更多信息,请扫描二维码联系我们!
电话:19962541230
邮箱:caogeyi@metahub.cn
